“If you torture the data long enough, Nature will confess...…”
We develop and apply advanced computational methodologies in bioinformatics of proteins and RNAs. These methods combine novel and existing algorithms drawn from machine learning, statistics, and bigdata analytics. Our projects range from analysis and prediction of bigdata in the order of 10 million proteins/RNAs and several thousands of organisms, through analysis of individual organisms (thousands of proteins/RNAs), disease models, to analysis of individual proteins/RNAs. Our research projects concern - Prediction and modeling of structure and function proteins and RNAs - Discovery and characterization of sequence-structure/disorder-function relationships in proteins - Binding of small ligands (including drugs), peptides, RNAs and DNA to proteins - Prediction and functional characterization of intrinsic disorder in proteins
We develop advanced translational systems that are used to convert complex (multi-system, multi-source and multi-format) clinical data into diagnostically- and prognostically-relevant observations. We collaborate with computer scientists, bioinformaticians, biochemists, structural biologists, biophysicists and clinicians to analyze a variety of such datasets that are relevant to several human diseases including cystic fibrosis, arthritis, and heart failure.
A sample of our projects follows.
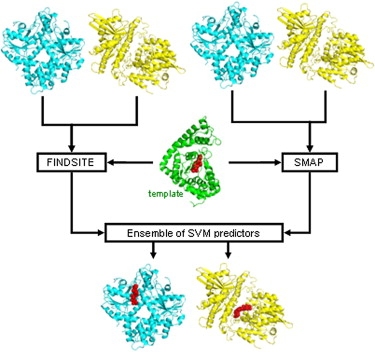
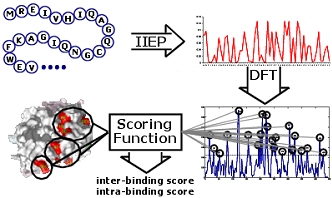
Prediction of binding sites has applications in rational drug design and in the discovery of protein-protein and protein-ligand interactions. We developed several machine learning methods that predict binding residues in the protein sequence and binding sites in the protein structures. Our efforts concentrate on predictions for:
We performed a comprehensive critical evaluations of in-silico predictors of the binding sites for biologically relevant organic compounds. [Structure 19:613-21, 2011] and for sequence-based prediction of DNA- and RNA-binding [Brief Bioinformatics DOI 10.1093/bib/bbv023, 2015]. This former contribution highlights the current developments in this area and positions our group at the forefront of this field. We are in the possession of unique and invaluable insights into building of the next generation predictors and we authored the most comprehensive benchmark dataset and results. We currently focus on the inverse binding site predictions, where knowledge of one or a few protein-ligand complexes is used to find other binding partners for this ligand on a proteomic (whole organism) scale. This work finds applications in discovery, characterization, and mitigation of side effects of clinically used and novel small molecule-based drugs and in drug repurposing. We published a cutting-edge inverse binding site predictor, ILbind, which provides favorable predictive performance on a large and diverse set of 35 compounds [Structure 20:1815-22, 2012] and comprehensively characterized novel interactions between human proteins and a widely used immunosuppressant Cyclosporin A [Bioinformatics 30(24):3561-66, 2014].
For years, scientists were convinced that proteins must fold into precise, rigid molecules to function correctly. This view is changing now. The intrinsically disordered proteins (IDPs) have at least some disordered (also called unfolded or highly flexible) regions and many of them carry out their function without ever fully folding into a rigid molecule. This flexibility is crucial for signal transduction, regulation of cell division, transcription, translation, phosphorylation, and many other cellular functions. The prevalence of disorder was also shown in human diseases including cancers, cardiovascular, neurodegenerative, and genetic diseases. Experimental annotations of IDPs are time consuming and difficult and thus computational methods that predict disorder from sequences have emerged as a viable alternative to investigate the disorder. These methods find numerous applications in functional and structural proteomics.
We have a history of excellence in this area. Our machine learning predictor, MFDp [Bioinformatics 26(18):i489-96, 2010], recently secured top three finish among 28 participants in the disorder prediction at the 10th worldwide CASP10 experiment [Proteins 82(Suppl 2):127-37, 2014]. We also developed first-of-its-kind predictor of disorder content, DisCon [BMC Bioinformatics 12:245, 2011], one of the most accurate methods for the prediction of disordered protein-binding regions, MoRFpred [Bioinformatics 28(12):i75-i83, 2012], high-throughput (genomic scale) predictor RAPID [BBA Prot Proteomics 1834(8):1671-80, 2013], the first-of-its-kind predictor of multiple disorder driven functions, disoRDPbind [Nucleic Acids Res 43(18):e121, 2015], that covers protein-, RNA- and DNA-binding, and we co-developed the repository of putative disorder [Nucleic Acids Res 41:D508-16, 2012]. Our methods are available as convenient webservers that enjoy a wide-spread use; we have processed requests from thousands of unique users from 80+ countries on all continents.
We also published one of the most comprehensive comparative reviews on the prediction of intrinsic disorder [Curr Prot Pept Sci 13:6-18, 2012] and performed characterization of the functional roles that the disorder plays. We just completed several ambitious, collaborative projects that analyzed the role of disorder in the proteome of the HIV-1 virus [Cell Mol Life Sci 69:1211-59, 2012], hepatitis C virus [Mol BioSystems 10:1345-1363, 2014] and dengue virus [FEBS J 282(17):3368-3394, 2015], in the nucleosome [Mol BioSystems 8:1886-901, 2012], in the programmed cell death peocess [Cell Death and Differ 20:1257-67, 2013], and in the ribosomal proteins [Cell Mol Life Sci 71(8):1477-1504, 2013]. As a recent highlight, we recently contributed to two landmark reviews on the role of disorder in protein complexes [Chemical Rev 114 (13):6806-43, 2014] and in viruses [Chemical Rev 114(13):6880-911, 2014]. We also published arguably the most comprehensive to date characterization of the abundance and functional roles of intrinsic disorder over all known domains of life [Cell Mol Life Sci 72(1):137-151, 2015] and on the abundance of MoRF regions [Mol BioSystems DOI: 10.1039/C5MB00640F, 2015].

MicroRNAs (miRNAs) are abundant and short endogenous noncoding RNAs made of 19-23 nucleotides that bind to target mRNAs, typically resulting in their degradation and translational repression. The fine-tuning of gene regulation in biological processes and disease pathways by these small RNAs recently attracted significant attention. We have developed advanced computational platform to characterize miRNA in large-scale deep-sequencing data and built novel methods for high-throughput machine learning-driven prediction of targets of known and novel miRNAs. Our platform to identify miRNA was featured in the project that investigated novel mechanisms of modulation of unfolded protein response in the endoplasmic reticulum stress [Science Signaling 7(329):ra54, 2014]. We also co-authored a novel method to predict miRNA targets in human genome, HiMiTar [Algor Mol Biol 3:16, 2008], and we recently completed an ambitious and comprehensive overview and assessment of computational predictors of microRNA targets in animals [Brief Bioinformatics 16(5):780-794, 2015]. We are currently working on a new generation of methods that aim to solve a challenging problem of high-throughput prediction of non-canonical targets of miRNAs..
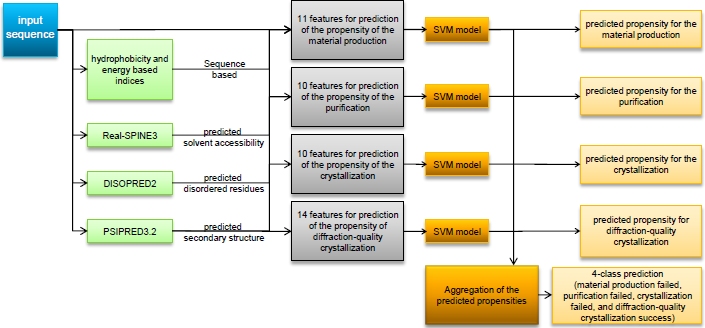
Structural Genomics (SG) is a multi-billion dollars international effort that aims at large-scale determination of 3D shapes of important biological macro-molecules with primary focus on proteins. One of the main bottlenecks in SG is the ability to produce diffraction quality crystals for the X-ray crystallography based protein structure determination, the dominant approach to solve protein structures. The SG pipelines allow for certain flexibility in target selection which motivates development of computational methods for the sequence-based prediction/assessment of the protein crystallization propensity. Over the last several years, we developed several novel machine learning methods for characterization and prediction of the crystallization propensity including CRYSTALP [Biochem Bioph Res Comm 355(3):764-9, 2007], CRYSTALP2 [BMC Struct Biol 9:50, 2009], MetaPPCP [Biochem Bioph Res Comm 390(1):10-15, 2009], CRYSpred [Prot Pept Lett 19(1):40-49, 2012] and most recently, PPCpred [Bioinformatics 27(13):i24-33, 2011] and fDETECT [Acta Crystal D D70:2781-2793, 2014]. Our methods are characterized by favorable success rates, when compared with state-of-the-art in the field, and they helped in identifying useful crystallization markers. The PPCpred is the first-of-its-kind method that provides insights and predictions to comprehensively cover the entire structure determination pipeline, including material production, purification, and crystallization.
Over the last two years, in collaboration with researchers at the Midwest Center for Structural Genomics, we completed an ambitious project that estimated current and projected levels of structural coverage in close to 1000 genomes from Eukaryota, Bacteria and Archaea [Acta Crystal D D70:2781-2793, 2014]. This work was recently featured in two popular science articles: "Unravelling the complexity of proteins" in the Dec 1, 2014 issue of the ScienceDaily (http://www.sciencedaily.com/releases/2014/12/141201100343.htm); and "Crystallography for Complete Proteomes" in the Dec 22, 2014 issue of BioTechniques (http://www.biotechniques.com/news/Crystallography-for-Complete-Proteomes/biotechniques-355553.html).
My group is a part of a large multidisciplinary team of researchers (Sherbrooke U., U. of Western Ontario, U. of Toronto, McGill U., and U. of Alberta) aiming at prevention, detection and management of bone loss in the context of rheumatoid arthritis and osteoarthritis. This project aims at (1) establishing osteoclastic markers related to presence and severity of bone and joint destruction; (2) determining whether these biomarkers are predictors of patient progression and responsiveness to different treatment regimens; (3) identification of new targets for development of antiresorptive therapies specifically aimed at arresting inflammation-induced bone loss. Our principal role in this project is to perform machine learning-driven and statistical analysis of an integrated structural, functional and molecular data to discover novel biomarkers and to design diagnostic and prognostic models. To date, the main outcomes are two high-quality journal articles (Bone 48:588-96, 2011; Arthritis Rheum 65(1):148-158, 2013).

Microtubules are the target of numerous antimitotic agents, including colchicine and antitumor drugs such as the taxanes, epothilones, and Vinca alkaloids. Unfortunately, these chemotherapeutic agents bind tubulin indiscriminately leading to the destruction of both cancerous and healthy cells. The differences within the β-tubulin isotypes expressed in a range of cell types may provide a foundation for the development of anti-tubulin drug derivatives with increased specificity for particular cancer cells. In collaboration with Dr. Tuszynski group at the Cross Cancer Institute in Edmonton, we investigate common patterns that lead to microtubule disruption and may provide a guide to the rational design of novel compounds that can inhibit microtubule dynamics for specific tubulin isotypes or resistant cell lines. Our aim is to develop computational methods that are capable of predicting relative differences in binding affinity to known drug binding sites within different tubulin isotypes. Relevant completed works include analysis of binding affinities of tubulin isotypes (J Mol Graph Model 27(4):497-505, 2008) and analysis of tubulin mutations and isotype expression in acquired drug resistance (Cancer Informatics 3:159-81, 2007). This work was featured in an article entitled "Zeroing In..." by Catherine Shaffer which was published in the December 2008 issue of Drug Design and Development magazine (the article can be found at www.dddmag.com/articles/2008/12/zeroing).
We developed several highly scalable algorithms that mine large/complex datasets to provide human-interpretable models. We focus on inductive supervised machine learning algorithms that generate simple models in terms of either production (if…then) rules, decision trees, and association rules. The models are flexible, i.e., they can be applied to a variety of input data formats (numerical, discrete and nominal), they can handle data containing noise and missing values, and are very fast to compute (in linear and log-linear time with respect to the number of input instances). These methods were used on datasets with hundreds of thousands up to several millions of instances (input vectors), which are typical to problems encountered in a proteome-wide analysis. Very few other existing inductive supervised learners can scale to problems of that size. Importantly, the models generated by our learners can be read and understood by users/domain experts and this is particularly important in biomedical domain where models have to be validated by the domain experts before they could be applied in a clinical setting. Our models provide a concise representation of the input data and can be used for predictive (both diagnostic and prognostic), annotative, and biomarker-discovery purposes. Algorithms developed in our lab include mi-DS (IEEE Tran. SMC B 2012, in print), ACRI (Data and Knowledge Eng 64(1):171-97, 2008), DataSqueezer (IEEE Trans. SMC B 36(1):32-53, 2006), MetaSqueezer (Proc ICMLA'04, 242-9), CLIP4 (Information Sciences 163(1-3):37-83, 2004), and CAIM (IEEE Trans. Knowledge and Data Eng 16(2):145-53, 2004). These methods were applied in practical contexts, e.g., MetaSqueezer and CAIM were used to analyze cystic fibrosis data in collaboration with Dr. Frank Accurso at the Children’s Hospital in Denver, and they were found useful by a wider research community. For instance, CAIM was implemented in a number of popular data analysis platforms, such as R and MATLAB, and received well over 350 citations, see http://scholar.google.com/scholar?hl=en&q=CAIM.

In spite of assuming a multitude of complex three-dimensional structures and bearing a wide range of biological functions proteins are characterized by simple and regular local folding patterns. The exponentially growing gap between the number of known protein structures (in dozens of thousands) and known protein sequences (in dozens of millions) motivates development of methods that identify structural and functional patterns using the known structures and apply the resulting models to predict/annotate these structural and functional characteristics on a proteome scale using the protein sequences as the inputs. Virtually all current prediction methods rely on sequence alignment that requires at least 30% sequence similarity between the query sequence and sequence(s) used to predict its structure. Proteins characterized by a lower, 20–30%, identity belong to the so called twilight zone. Over 95% of sequence pairs in the twilight zone have different structures (and therefore functions), which presents a considerable challenge for the existing prediction methods. We proposed a wide array of novel high-throughput methods that predict various structural and functional characteristics of the twilight-zone proteins based on their sequences. These models are built based on a hierarchical approach in which we utilize the protein sequence to predict certain relevant lower-level structural/functional characteristics which are aggregated and fed into nonlinear models, such as Support Vector Machines, to address characterization, prediction and analysis of higher-level characteristics. We developed a number of state-of-the-art predictors of
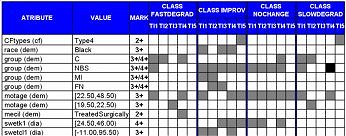
Lung disease in cystic fibrosis (CF) is the primary cause of early morbidity and mortality in affected patients, and is the focus of an extraordinary research effort worldwide. In spite of strong research push concerning variations in the decline in pulmonary function in CF patients, little is understood about the contributing risk factors. This project, which involved analysis of a large and complex clinical database, allowed identifying new and confirming known factors that may affect the progression of this disease. The database of over 850 patients was analyzed using a novel data mining system, MetaSqueezer, and our research demonstrated an important role of sweat electrolytes in CF.
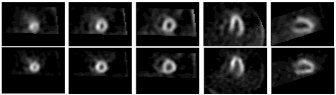
Modern medicine generates huge amounts of image data that can be analyzed and processed only with the use of specialized computer software. Imaging techniques like SPECT, PET, and MRI can generate gigabytes of data per day. There are many advantages of computerized analysis of data over human analysis: lower price, shorter time, automatic recording of analysis results, consistency, relatively inexpensive re-use of previous solutions. Our goal was to create a computer system that is able to semi-automate the cardiac Single Proton Emission Computed Tomography (SPECT) myocardial perfusion diagnostic process. We automatically computed a set of partial diagnoses for regions of the left ventricle (LV) muscle, as well as the overall diagnosis, which describes perfusion of the entire LV cardiac muscle. A database consisting of over 250 cleaned patient SPECT images (about 3000 2D images), accompanied by clinical information and physician interpretation was used to create a new user-friendly system for computerizing the diagnostic process (Artif Intel Med 23(2):149-169, 2001).