ncMirTar Help and Tutorial
This page explains how to use non-canonical microRNA target (ncMirTar) predictor to query targets of a known microRNA and to predict targets of a new microRNA, and how to understand the predicted result.
Search targets from the database
ncMirTar database provides a quick access to the pre-computed non-canonical microRNA targets. The users can retrieve the targets from three points of view according to their needs.
-
Input both microRNA and mRNA to check the propensity of user-interested microRNA-mRNA interaction

1 Input microRNA id, following the nomenclature of miRBase.
2 Input mRNA gene name or transcript accession id, following the nomenclature of Genbank and RefSeq.
3 Input your email address. Searching job will be queued and may wait until the server is available.
4 Press Search database to submit the job.
-
Input microRNA only to retrieve all targets predicted by this microRNA
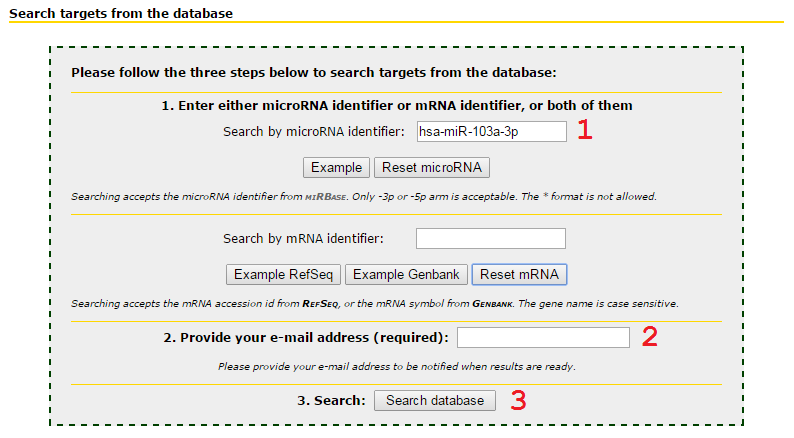
1 Input microRNA id, following the nomenclature of miRBase.
2 Input your email address. Searching job will be queued and may wait until the server is available.
3 Press Search database to submit the job.
-
Input microRNA only to retrieve all targets predicted by this microRNA
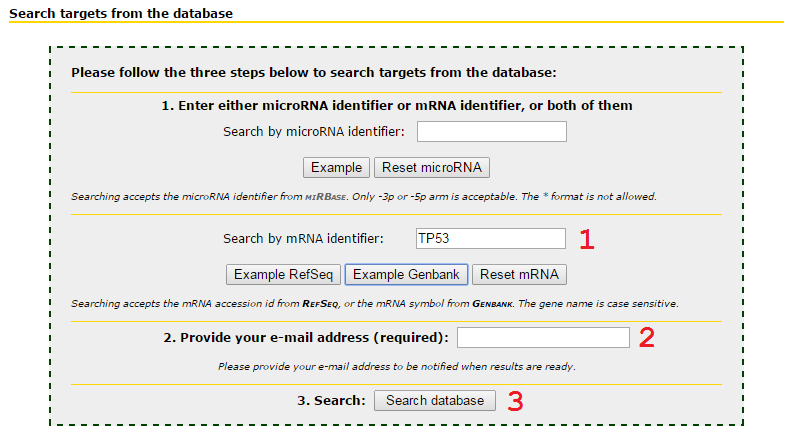
1 Input mRNA gene name or transcript accession id, following the nomenclature of Genbank and RefSeq.
2 Input your email address. Searching job will be queued and may wait until the server is available.
3 Press Search database to submit the job.
Predict targets using ncMirTar
ncMirTar web server offers to predict non-canonical targets of new microRNAs. The users can retrieve the targets of their microRNAs by inputting the microRNA sequences.
-
Input the species and sequence of interested microRNA to predict non-canonical targets of the given microRNA sequence
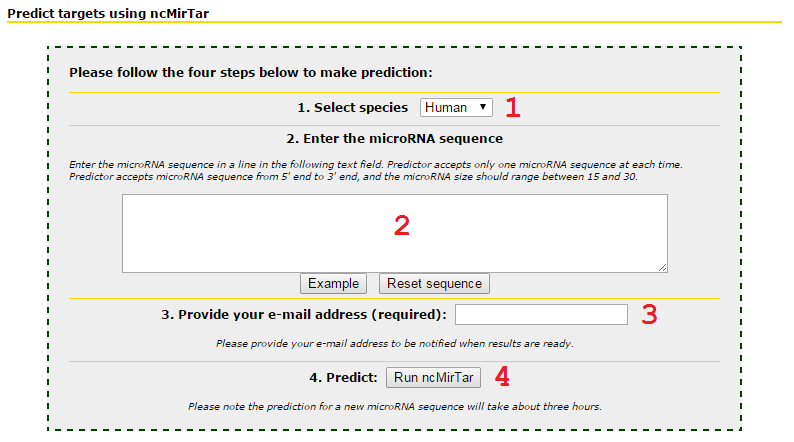
1 Choose species: either Human or Mouse.
2 Input microRNA sequence from 5' end to 3' end. It doesn't mater if you type captial letters or small letter. Prediction will not run when sequences have letters other than ACGTU.
3 Input your email address. Prediction takes up to three hours. The job will be queued and may wait until the server is available.
4 Press Run ncMirTar to submit the job.
Results from searching the database
When the input microRNA and mRNA ids are both valid, the result page will show 1) the queried microRNA-mRNA interactions or 2) No result meets the criteria of the query.
When either the input microRNA or mRNA id is not valid, user may expect four possible warning listed 3-6.
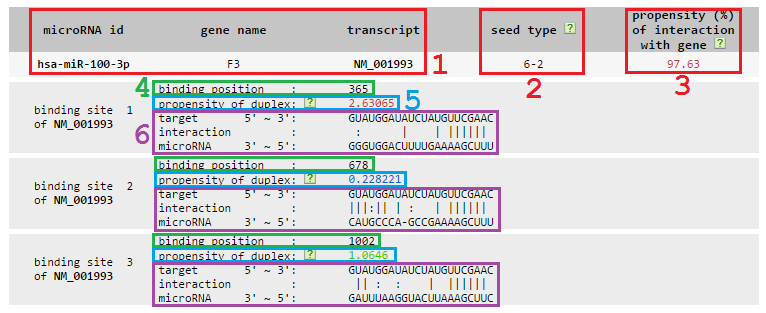
1 microRNA id, mRNA gene name and mRNA transcript accession id.
2 Seed type, two digits are connected by hyphen. The first digit means the maximum Watson-Crick base pairs in the seed region, and the second one gives the starting position of the maximum matches from the 5' end of microRNA.
3 Propensity of the microRNA-mRNA interaction. We sum up all propensities of the miRNA-target duplexes in this mRNA, rank this summed propensity among all propensities in the genome and output the normalized ranking as the propensity of the microRNA-mRNA interaction. The range of the normalized ranking is between 0 and 100, with that values higher than 70 are high probability and shown in red, that values lower than 50 are low probability and shown in blue, and that values in between are median probability and shown in green.
4 Binding position of the miRNA binding site on the mRNA, where the origin of the coordinate is the start of 3' UTR.
5 Propensity of the microRNA-target duplex. This value is the raw score outputted from SVM models. We remove the duplexes whose scores are below zero from this database. Scores given by 6 mer and 5 mer models are independent. Thus, we give two sets of thresholds to define their high/median/low propensity. For 6 mer model, values that are higher than or equal to 1.721 are high probability and in red, that are lower than 0.889 are low probability and in green, and that are in-between are median probability and in blue. For 5 mer model, values that are higher than or equal to 3.042 are high probability and in red, that are lower than 1.326 are low probability and in green, and that are in-between are median probability and in blue.
6 Detailed binding information consists of three lines: 1) target sequence bound by the given microRNA from 5' end to 3' end; 2) interaction between the microRNA and its target, '|' for Watson Crick base pairs, ':' for GU wobbles and ' ' for mismatches; 3) microRNA sequence from 3' end to 5' end.
- No target is found
- species does not exist
- microRNA does not exist
- transcript does not exist
- gene does not exist
Results from ncMirTar predictor
The server will first compare the query sequence with all microRNA sequences in our database. If the query sequence matches one of known microRNAs, the server will return the predictions of the matched microRNA from our database; otherwise the server will show the most similar known microRNA sequence(s) at E-value<0.001. Users can 1) consult the predictions of one of the similar microRNAs or 2) still predict targets of the query microRNA sequence.
- Results of alignment
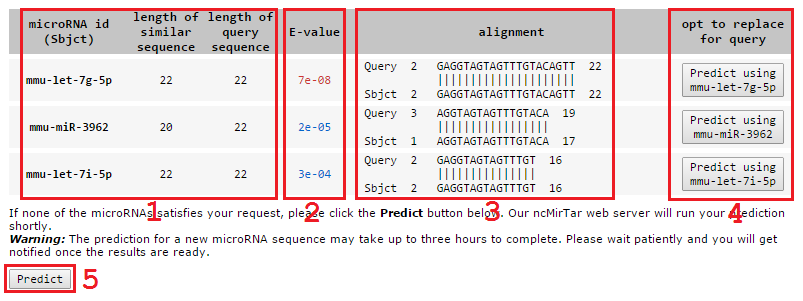
1 microRNA id and size of the silimar microRNA and the query sequence.
2 Expect value from blastn describes the number of hits that users can expect to see by chance. The lower the E-value, the more significant the match is. We already set the E-value threshold as 0.001 when running the blastn program, we also denote the extreme similar microRNAs by coding their E-values in red and the rest E-values in blue. The cut-off of extreme similarity is 1e-5.
3 Detailed alignment between the query sequence and the similar microRNA.
4 Click one of these buttons to view the predictions of the similar microRNAs.
5 Click the Predict button to run ncMirTar web server to predict the targets of the query microRNA.
- Results of target predictions of the new microRNA sequence
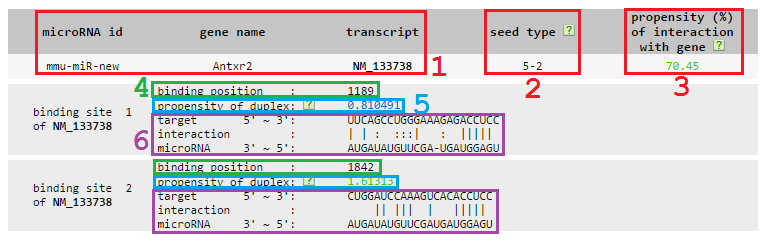
1 microRNA id, mRNA gene name and mRNA transcript accession id.
2 Seed type, two digits are connected by hyphen. The first digit means the maximum Watson-Crick base pairs in the seed region, and the second one gives the starting position of the maximum matches from the 5' end of microRNA.
3 Propensity of the microRNA-mRNA interaction. We sum up all propensities of the miRNA-target duplexes in this mRNA, rank this summed propensity among all propensities in the genome and output the normalized ranking as the propensity of the microRNA-mRNA interaction. The range of the normalized ranking is between 0 and 100, with that values higher than 70 are high probability and shown in red, that values lower than 50 are low probability and shown in blue, and that values in between are median probability and shown in green.
4 Binding position of the miRNA binding site on the mRNA, where the origin of the coordinate is the start of 3' UTR.
5 Propensity of the microRNA-target duplex. This value is the raw score outputted from SVM models.We remove the duplexes whose scores are below zero from this database. Scores given by 6 mer and 5 mer models are independent. Thus, we give two sets of thresholds to define their high/median/low propensity. For 6 mer model, values that are higher than or equal to 1.721 are high probability and in red, that are lower than 0.889 are low probability and in green, and that are in-between are median probability and in blue. For 5 mer model, values that are higher than or equal to 3.042 are high probability and in red, that are lower than 1.326 are low probability and in green, and that are in-between are median probability and in blue.
6 Detailed binding information consists of three lines: 1) target sequence bound by the given microRNA from 5' end to 3' end; 2) interaction between the microRNA and its target, '|' for Watson Crick base pairs, ':' for GU wobbles and ' ' for mismatches; 3) microRNA sequence from 3' end to 5' end.